Battling COVID-19; A New Model of Cytokine Release Syndrome
Under certain circumstances in patients, the human immune system can spin out of control and become highly toxic, resulting in cytokine release syndrome (CRS). CRS has been observed in certain autoimmune diseases (Grom et al. 2016), during highly infectious diseases like COVID-19 (Zhang et al. 2020), and following immune-enhancing treatments that include monoclonal or bispecific antibodies, or CAR T therapies (Shimabukuro-Vornhagen et al. 2018). CRS, which can be deadly, has been notoriously difficult to study and for which to develop novel treatments. However, encouraging new data indicates that many aspects of human CRS can be modeled in immunodeficient NSG mice engrafted with human peripheral blood mononuclear cells (PBMCs). This model provides hope that new immuno-modulatory therapies can be safely developed and tested before clinical trials.
Maintaining a fine balance
Human immunity is a highly intricate system comprised of unique cell populations capable of communicating and interacting with one another, as well as with the tissues around them, via a complex array of soluble and membrane-bound ligands and receptors. The primary function of the immune system is to recognize and eliminate foreign invaders while also limiting damage to host cells and tissues. The immune system has evolved complex mechanisms to protect healthy “self” cells, while identifying and killing compromised or infected cells. The mechanisms are carefully activated and, once the threat has been neutralized, the responses are then de-activated.
Unfortunately, the stability of the immune system can be lost, causing a range of systemic toxicities, even death. Adaptive and innate immune cells can become overstimulated, releasing inflammatory cytokines that promote further cytokine production from endothelial cells and fibroblasts. The hypersecretion of these factors is referred to as cytokine release syndrome (CRS), which can lead to vascular permeability, pulmonary edema requiring ventilation, liver damage, cytopenia, cardiomyopathy, and neurotoxicities. While CRS can occur in more than one disease setting, it typically begins with a powerful immune stimulus (Grom et al. 2016, Shimabukuro-Vornhagen et al., 2018, Zhang et al. 2020) such as an infectious disease like COVID-19.
CRS and COVID-19
Certain infectious agents — and particularly viruses — are capable of eliciting strong immune reactions that lead to CRS. In the case of the novel coronavirus SARS-CoV-2, the virus gains entry into cells and initiates its viral replication process via angiotensin-converting enzyme-2 (ACE2) receptors on the surface of epithelial cells in the airway, leading to the development of COVID-19. A subset of patients with COVID-19 will progress into respiratory distress following destruction of alveolar epithelial cells and the hyperactivation of both adaptive and innate immune cells. In these cases, macrophages and epithelial cells are extensively activated in a large-scale, pro-inflammatory response, leading to CRS (Zhang et al., 2020, Vabret et al. 2020).
Modeling Human CRS in the Mouse
Finding a suitable or appropriate mouse model is vital to understanding complex syndromes, such as CRS, and required for development of new treatments to enter into clinical trials. The Innovation and Product Development Group at The Jackson Laboratory (JAX) set out to develop a model to address the gap for this much needed area. The new model would have to meet a few requirements including engrafted human immune cells and the ability to reliably create CRS using agents known to cause CRS clinically while also demonstrating a lack of CRS when introducing clinically safe immuno-modulators. The model should allow both the screening of new drugs for CRS susceptibility and evaluation of new therapies capable of suppressing CRS.
Human PBMCs can be reliably engrafted into highly immunodeficient NSG™ mice (stock # 005557). Human PBMC-engrafted mice were tested for human immune cell-mediated CRS response by treating them with a wide range of monoclonal antibodies (mAb), some of which are clinically known to induce CRS (CRS positive group) and others that do not. The CRS positive group included a biosimilar to TGN1412 (anti-CD28 superagonist mAb), Muromonab (OKT3, anti-CD3 mAb), an alternate anti-CD28 mAb and Thymoglobulin (ATG). The group of drugs known to have no or minimal CRS response included Pembrolizumab (anti-PD1 mAb) and Rituximab (anti-CD20 mAb). Mice were treated six days after human PBMC engraftment and serum was collected at hourly intervals after treatment to perform quantitative human cytokine analysis. All mAbs in the CRS positive control group exhibited hyper-elevated levels of human IFN-ɣ, TNF-α, IL-2, IL-4, IL-6, and IL-10. Cytokine levels in the CRS negative group were similar to those in the PBS-treated control group. In order to further ensure the observed responses were drug mediated, TGN1412 analogue was tested at three doses and compared to the anti-CD28 positive control. TGN1412 analogue demonstrated a clear escalating dose-response of increased cytokine production across all time points examined (figure 1).
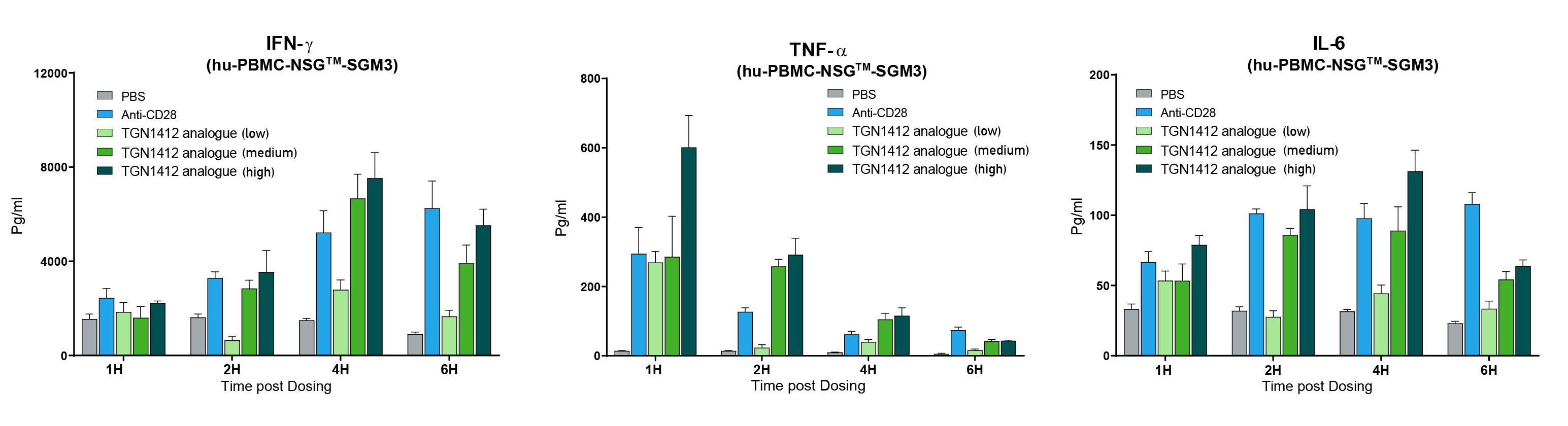
Figure 1. TGN1412 analogue (anti-CD28 superagonist) mediated CRS dose response. Human PBMC engrafted NSG™-SGM3 recipient mice (stock# 013062) were treated with anti-CD28 mAb positive control or TGN1412 analogue at low, medium or high dosing. Human IFN-ɣ, TNF-α, and IL-6 were measured quantitatively at 1, 2, 4, and 6 hours post treatment. Increasing doses of TGN1412 analogue showed a progressively increased cytokine release response.
The next question was whether PBMC donor-specific differences in drug response could be observed, mimicking what is observed clinically. Cohorts of mice were engrafted with nine different human PBMC donors, and each donor cohort was tested with PBS, OKT3, or anti-CD28. In mice treated with OKT3, 7 out of 9 donors displayed a severe IFN-ɣ CRS response, 5 showed a severe IL-6 response and all exhibited a strong TNF-α response (figure 2). Donors G and H showed a milder response. When treated with anti-CD28, 2 out of 9 donors showed a severe IFN-ɣ response. Collectively, the data described in the experiments above demonstrate that specific mAbs are capable of initiating a CRS response in this model and that the response is both dose and PBMC donor dependent.
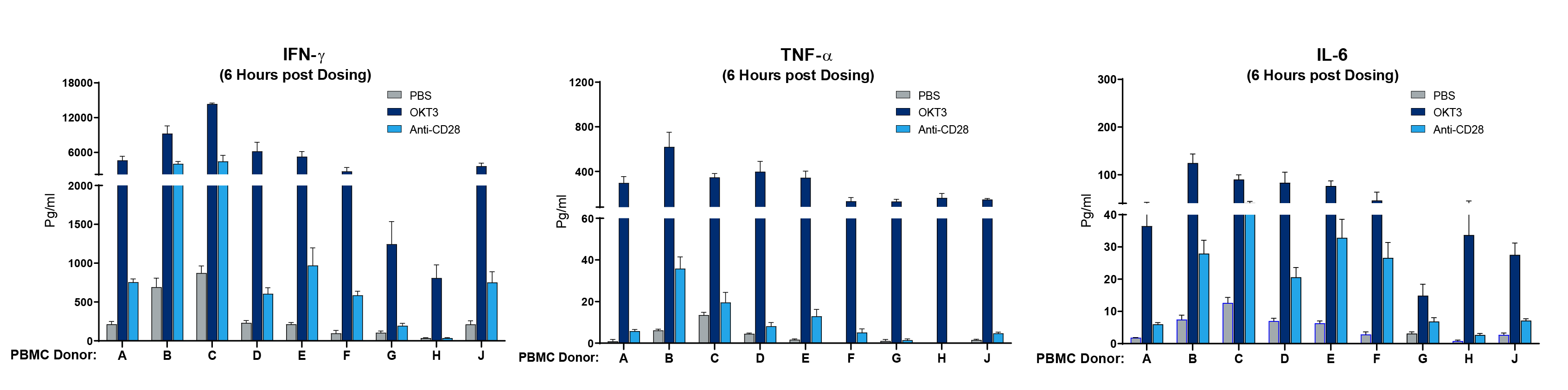
Figure 2. Human PBMC donor specific differences in magnitude of CRS response. Human PBMC from 9 different donors (A-J) were engrafted into NSG recipient mice (stock# 005557) and treated with Muromonab (OKT3, anti-CD3 mAb) or anti-CD28 mAb. Human IFN-ɣ, TNF-α, and IL-6 were measured quantitatively 6 hours post treatment. Each donor shows a quantitative difference in cytokine response to each drug tested.
Preclinical Treatment of Human CRS
The above data is a small sample of experiments performed to demonstrate human CRS can be successfully modeled in immunodeficient mice. Relative to COVID-19 mediated CRS, there is an urgent need to rapidly find treatments that can help assist in reducing immune-mediated pathologies. Advantages to the model described here include the short timeline needed to set up and run an experiment, human immune cell specific responses, and the fact that the model does not require complex ABSL3 infectious disease capabilities. As a further proof-of-principle for using the model in preclinical treatment, the JAX Innovation and Product Development group used two human PBMC donors (noted A&B) to engraft cohorts of NSG™ mice, and the CRS was initiated using anti-CD28 or OKT3. Preclinical testing was performed by treating the induced CRS response with the clinically approved anti-TNF-α mAb drug, Humira (figure 3).
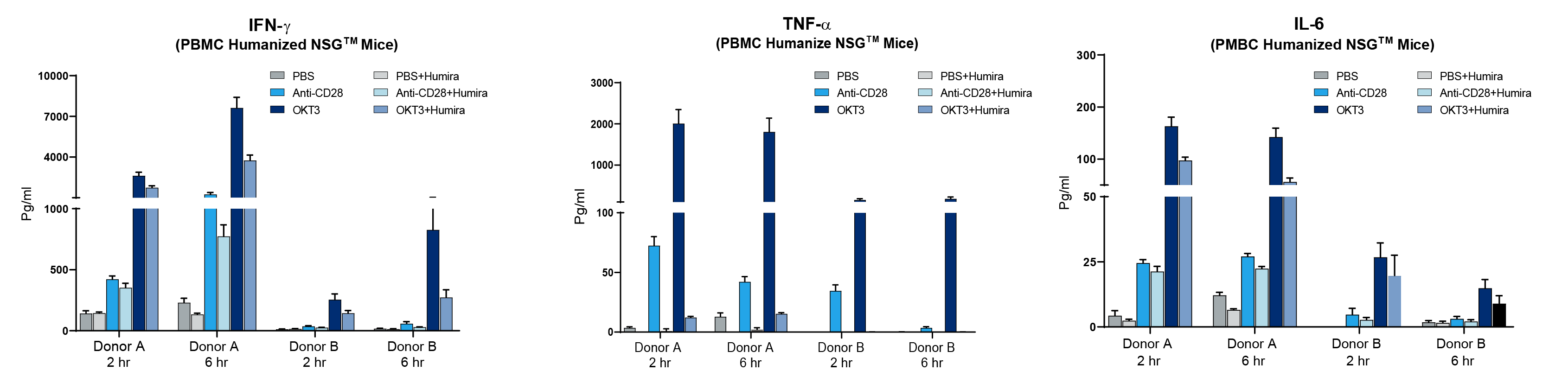
Figure 3. Humira (anti-TNF-α mAb) diminishes CRS response in PBMC humanized mice. Two different human PBMC donors (A&B) were engrafted into NSG recipient mice (stock# 005557) and treated with Muromonab (OKT3, anti-CD3 mAb) or anti-CD28 mAb alone or combined with Humira. Human IFN-ɣ, TNF-α, and IL-6 were measured quantitatively in peripheral blood at 2 and 6 hours post treatment. Humira nearly eliminated TNF-α and diminished IFN-ɣ and IL-6.
Donor A exhibited a strong CRS response with anti-CD28 or OKT3 while donor B showed amilder response. Humira treatment, as expected, led to a dramatic decrease in human TNF-α when examining the peripheral blood of mice engrafted with either donor. Humira treatment also reduced IFN-ɣ and IL-6 compared to mice treated with anti-CD28 or OKT3 alone. While Humira did not completely mitigate CRS in this model, it clearly demonstrated that a human specific therapeutic agent could neutralize its target and diminish other cytokines associated with CRS. Additional parameters that can be used to examine effectiveness of preclinical response (not shown here) include changes in body weight, disease activity index scoring, survival, and histopathology.
As JAX is committed to developing the most innovative products and services, we are pleased to now offer a cytokine release platform. This platform provides rapid, accurate assessments of cytokine release markers for preclinical toxicity and anti-inflammatory studies of novel therapeutics, detecting variability across human blood cell donors with greater sensitivity than traditional methods. Visit the JAX Breakthrough lab page to learn more about this pioneering model.
Recommended Resources:
Mouse Model for COVID-19 Research
References
Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, Kochanek M, Böll B, von Bergwelt-Baildon MS. “Cytokine release syndrome“. J Immunother Cancer. 2018 Jun 15;6(1):56. doi: 10.1186/s40425-018-0343-9. PMID: 29907163
Grom AA, Horne A, De Benedetti F. “Macrophage activation syndrome in the era of biologic therapy“. Nat Rev Rheumatol. 2016 May;12(5):259-68. doi: 10.1038/nrrheum.2015.179. Epub 2016 Mar 24. PMID: 27009539
Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. “The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality“. Int J Antimicrob Agents. 2020 Mar 29:105954. doi: 10.1016/j.ijantimicag.2020.105954. Online ahead of print. PMID: 32234467
Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, Levantovsky R, Malle L, Moreira A, Park MD, Pia L, Risson E, Saffern M, Salomé B, Esai Selvan M, Spindler MP, Tan J, van der Heide V, Gregory JK, Alexandropoulos K, Bhardwaj N, Brown BD, Greenbaum B, Gümüş ZH, Homann D, Horowitz A, Kamphorst AO, Curotto de Lafaille MA, Mehandru S, Merad M, Samstein RM; Sinai. “Immunology of COVID-19: Current State of the Science.” Immunology Review Project. Immunity. 2020 May 6;52(6):910-41. doi: 10.1016/j.immuni.2020.05.002. Online ahead of print. PMID: 32505227