Evidence-based selection of the starting dose in first-in-human clinical trials using humanized mouse models

The dose selection for first-in-human (FIH) clinical trials relies on data obtained by a combination of in vitro and in vivo studies designed to evaluate the efficacy, toxicity, and stability of the investigational drugs. The entirety of the preclinical data is integrated with historical information on similar compounds to predict a safe dosing regimen.
Unfortunately, the preclinical development of antibodies and most biologics suffers from the lack of predictive in vivo models. The main characteristic of these molecules, namely their specificity, means that they are inactive in virtually all non-primate models because of the lack of appropriate targets. The current shortage of non-human primates (NHPs), recognized by the regulatory agencies (1), makes it even harder for drug developers to generate the preclinical pharmacokinetic (PK), safety, and efficacy data required to file investigational new drug applications. The lack of these data makes it challenging to define the dosing regimen for FIH clinical studies (2).
Humanized mice represent a new generation of tools for the preclinical development of drugs recognizing human-specific targets. In the past few years, the development of these models has provided preclinical researchers with several options for reducing the number of NHPs used in the drug development process.
Predict the serum stability of an Fc- and albumin-conjugated test article in patients using genetically humanized mouse models.
The serum half-life of most circulating large molecules, including antibodies and albumin, is actively regulated by the endothelial cells. The circulating proteins are constantly endocytosed and targeted for degradation in the lysosomes by the cells lining the blood vessels. Fc-containing molecules (such as IgGs) and albumin bind to the neonatal Fc receptor (FcRn) and escape degradation to be recycled in circulation. Since species-specific sequences in the Fc portion of antibodies and in albumin mediate the interaction with FcRn, it is impossible to extrapolate the stability of an Fc- or albumin-conjugated drug using the classical mouse or rat models (Figure 1a). To address this issue, Derry Roopenian and collaborators at the Jackson Laboratory generated several genetically humanized mouse models replacing the expression of the mouse FcRn with its human counterpart (3 And Figure 1b). Years of validation and numerous publications by independent groups confirmed that the clearance of Fc- and albumin-conjugated therapeutic molecules in these models reflects the one observed in NHPs and humans. These FcRn humanized mice allow researchers to investigate the effects of molecular engineering on the stability of their drug candidates and to predict their serum half-life in patients.


Figure 1. Comparison of the clinical serum half-life of three therapeutic antibodies with the corresponding half-life in a) WT mice or b) FcRn humanized (Tg32) mice, respectively. The R-squared of the linear regression model is shown in the graph, indicating a strong correlation between the human PK of these molecules and that in FcRn humanized mice but not in WT animal.
Testing the efficacy/toxicity dose response of human-specific biologics using immune system-humanized mouse models.
Biologics such as therapeutic monoclonal antibodies do not recognize their targets in most animal models. The lack of relevant in vivo models limits the ability to perform dose-response experiments that could guide the process of defining appropriate clinical regimens. In many cases, researchers move to the clinic without the in vivo data required to select a safe and efficacious starting dose, especially for immmunomodulatory combination therapies. As a result, clinical scientists rely on historical data to define the dosing regimen of FIH studies, which is especially problematic for molecular entities with innovative formats. They often need to adopt cumbersome dose-escalation approaches based on the minimally-anticipated biological effect level (MABEL) calculated using suboptimal in vitro preclinical assays. Even when NHP data are available, the lack of the tumor-specific antigen in this system makes these studies less translationally relevant. As a result, FIH clinical studies for immunoactivating biologics have an intrinsic risk of serious adverse events due to the absence of information on safe dosing, as exemplified by TGN1412 (4) and, more recently, by the Regeneron REGN1919 bispecific antibody (5).
Highly immunodeficient mice engrafted with human peripheral blood mononuclear cells (PBMCs) support the engraftment of a functional immune system (6) and can be used to study the systemic effects of immunostimulatory drugs. Crucially, it is possible to implant tumors in these humanized mice. This configuration allows researchers to test the antitumoral response mediated by immunoactivating molecules and to assess their acute toxicity effects (e.g., CRS) in the presence of both the tumor and the immune system. A straightforward application of this humanized system is the assessment of the potential immunotoxicity of drugs activating the immune system. The CD28 superagonist TGN1412 is a well-known example of a drug that did not show any overt toxicity in preclinical assays but almost killed the volunteers participating in a phase 1 clinical trial. As seen in Figure 2, the JAX assay based on PBMC-humanized mice detects both the cytokine release induced by TGN1412 and the organ toxicity caused by the unchecked activation of the immune system, confirming the translational value of the system. Since the response is dose-dependent, these data could be used to inform the selection of the starting dose in a human clinical trial.
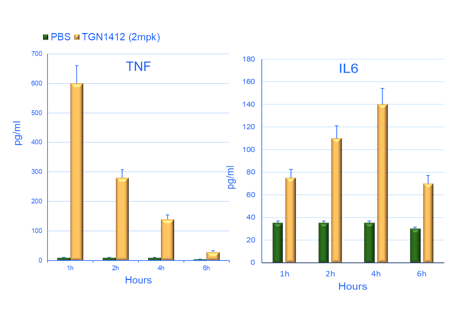
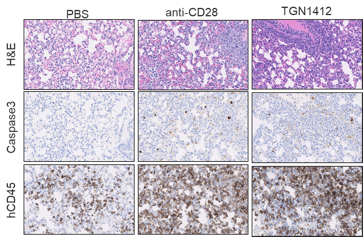
Figure 2. Effect of the treatment of PBMC humanized mice with the CD28 superagonist drug TGN1412. An example of the time course of two pro-inflammatory cytokines is shown in a) and b). The identity of the cytokine is shown above the bars. The horizontal axis shows the hours post-drug treatment, while the vertical axis represents the pg/ml detected in the blood of the treated animals. The green bars correspond to the negative control (injection of the vehicle). The panels on the right (c) illustrate representative examples of lung tissue sections collected from humanized mice treated with the vehicle (PBS), a positive control (anti-CD28) and TGN1412 10 days post-treatment. The stainning is indicated on the side of the panels. H&E (hematoxylin and eosin) staining shows numerous tissue-infiltrating cells. Human CD45 staining confirm that the cells infiltrating the tissue are human immune cells. Caspase 3 staining reveals active apoptosis in the tissue. A pathology analysis confirms the tissue damage induced by TGN1412 (not shown).
A more complex and potentially more useful experimental design allows the exploration of the relationship between efficacy and toxicity of immunoactivating biologics. By injecting luciferase-labeled tumor cells in PBMC-humanized mice and treating the animals with different concentrations of the drug candidate, it is possible to establish dose-response toxicity and efficacy curves that can guide clinical scientists in the selection of a safe and efficacious FIH starting dose (Figure 3).
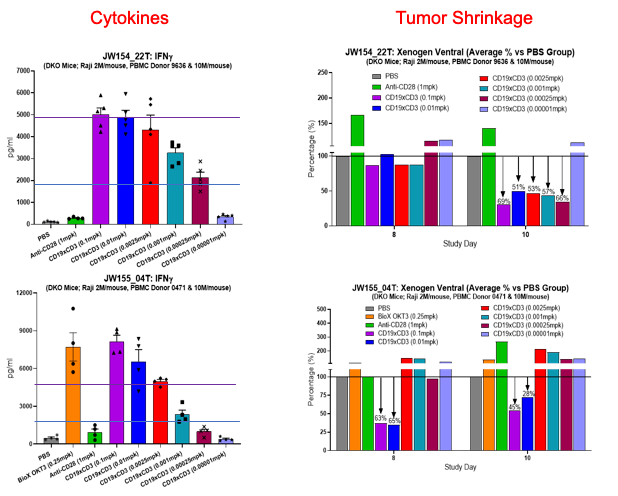
Figure 3. Anti-tumor efficacy and acute toxicity in mice humanized with PBMCs from different donors (indicated on the left) of a bispecific antibody recognizing CD19 and CD3. The assay and experimental conditions are shown above the graphs. a and c): the treatment with the CD3 engager results in a strong dose-dependent cytokine release. The test article and its concentration are shown on the horizontal axis. PBS is the negative control. Anti-CD28 and OKT3 are the positive controls. b and d): the residual luminescence, shown as percentage of the one observed in control-treated animals, indicates the persistence in the animals of luciferase-labelled tumor cells eight and ten days after the drug treatment. A number lower than 100% suggests tumor growth inhibition. The lack of a linear correlation between toxicity and efficacy allows the selection of a dose where the efficacy is still retained and the toxicity is not problematic. Note that the two donors have different toxicity/efficacy profiles, reflecting the clinical variability seen in patients.
By running a simple PK/bioavailability study in the same model and integrating the data on the same molecule using the FcRn platform (see the previous section), clinical scientists can extrapolate the stability of their molecule in patients and rationally identify a dosing regimen.
Real-world complexity in preclinical studies
A significant limitation of most preclinical studies lies in the genetic diversity clinical researchers face when treating a human population. From the practical point of view, patients respond very differently to the same drug, with reactions varying from none to severe adverse events. PBMC-humanized mice can capture this diversity. The same drug treatment of mice humanized with different donors can elicit very different responses, mirroring the clinical reality (Figure 4). Again, this crucial information can be integrated into determining the first dose of a clinical trial to ensure that even high responders will not be at risk of incurring severe adverse events.


Figure 4. The cytokine release induced by immunoactivating drugs in mice humanized by PBMC from different donors mirrors the variability observed in the clinic. The horizontal axis shows the different donor ID. The release of INFy and IL10 is shown on the left and right panes, respectively. The different colors of the bars refer to the specific treatment (see graph legends).
Conclusion
At this point, there is no defined preclinical path to select the first dose in an FIH clinical trial for antibody-based drugs. The vast diversity in the nature of the target antigens and the format of the drug candidates imply a lack of reliable preclinical or historical data for the selection of a safe starting dose in escalation studies. We suggest that the combination of genetically and immune system-humanized mice can provide preclinical researchers using the MABEL approach with increased confidence in predicting the start dose for their clinical studies. As a future development, it is not impossible to conceive that the PBMC of the very patients that will be treated can be used to predict their response to the specific immunoactivating drug, opening the path to personalized treatment plans.
References
- https://www.fda.gov/media/155950/download
- Saber et al., (2017). An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul Toxicol Pharmacol ;90:144-152. doi: 10.1016/j.yrtph.2017.09.001
- Petkova et al.,(2006). Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 18(12):1759-69 doi: 10.1093/intimm/dxl110.
- Suntharalingam et al., (2006). Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 355(10):1018-28. doi: 10.1056/NEJMoa063842
- https://endpts.com/regeneons-star-bispecific-is-linked-to-2-deaths-in-a-small-study-which-was-no-help-for-its-q1-call
- King M, et al., (2007). Development of new-generation HU-PBMC-NOD/SCID mice to study human islet alloreactivity. Ann N Y Acad Sci. 1103:90-3. doi: 10.1196/annals.1394.011.