Using RNA sequencing and other technologies to aid in variant interpretation
Malik is your 6-year-old patient with autism and global developmental delay. DNA testing did not identify a pathogenic variant (mutation) using a multi-gene neurology panel. However, a variant of uncertain clinical significance (VUS) was identified in a gene associated with a condition that includes autism among the observed clinical features. This result is frustrating, and you wonder if there is a way to clarify his risk of having this condition.
Genomic testing is moving beyond DNA testing.
The majority of pathogenic variants are identified by DNA analysis. However, for a discrete number of patients whose results are uninformative, it may be possible to get a more definitive answer about the consequences of a variant using additional methods like RNA sequencing, protein analysis and predictive or functional studies.
If your clinical and family history assessment make you suspicious of a genetic condition, DNA testing is currently still the first step to investigate this possibility. Results may be:
- positive, where a pathogenic variant adversely affecting gene function is identified;
- negative, where either benign or no variants are identified; or
- VUS, where the effect of the variant on gene function is unknown.
In the past, a result classified as a VUS would have been uninformative for clinical decision-making. While DNA testing may have stopped here, further testing is possible. RNA testing looks for changes in gene expression, and protein analysis assesses structural changes in the protein product of the gene. Employing these additional investigative techniques might provide additional information about a variant.
RNA sequencing and gene expression
RNA sequencing is beginning to show promise for understanding how a DNA variant may disrupt the normal transcription to RNA, post-transcriptional modifications, or the subsequent protein structure.
Currently, 10% of variants identified through genomic testing are classified as VUS in a widely used repository called ClinVar (Ittisoponpisan 2019). Of these, a small but significant number are in splice sites. For example, 11% of VUS in hereditary cancer genes are splice site variants (Karam 2019). When looking solely at VUS that are classified as “variant, likely pathogenic,” the proportion of splice site variants is much greater, at 55% (Karam 2019).
But what are splice sites? Genes contain both coding (exon) and non-coding (introns) sections that are transcribed into mRNA. This mRNA is processed, cutting out the non-coding portions and splicing together the remaining coding portions. Pathogenic variants can disrupt the splice sites at either end of the exon, create novel splice sites, reveal cryptic splice sites, or disrupt regulatory regions deep within the introns. All of these mechanisms negatively affect expression of the gene.
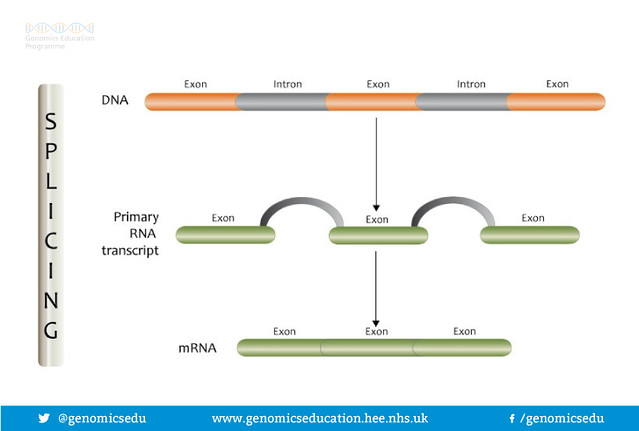
RNA Splicing
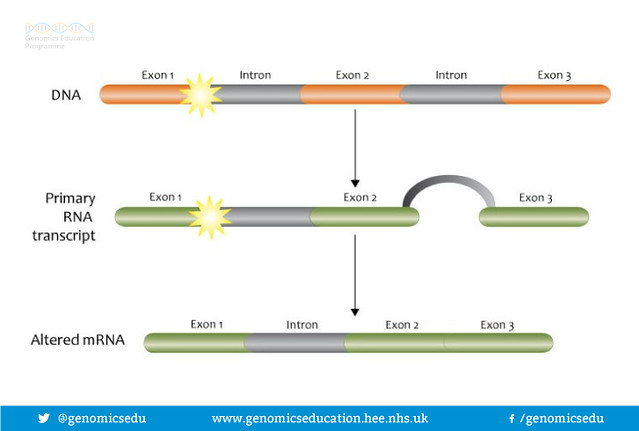
Splice site mutation
Lee (2020) and Frésard (2019) demonstrated clinical utility in adding RNA sequencing to genomic testing for cohorts of individuals with undiagnosed Mendelian disease, increasing the diagnostic yield by 18% and 7.5%, respectively. Landrith (2020) demonstrated in increase of 9.1% in the detection of pathogenic variants in 18 hereditary cancer genes. Karam (2019) was able to reclassify 6.2% VUS to pathogenic. Because of this, labs are beginning to offer paired simultaneous DNA-RNA testing or request additional samples for RNA sequencing.
Other complimentary techniques also help determine whether variants have clinical significance.
Protein analyses examine the primary structure (amino acids) as well as the secondary folded structure, more complex tertiary structures, and protein interactions. The predicted effects of a variant on functional areas of the protein such as binding sites for DNA or other proteins can also be analyzed. In silico modeling uses computational methods to simulate real biological processes in a virtual environment. Functional analyses use stem cells and other biological systems in vitro. And finally, family studies, which examine whether a variant tracking with affected relatives is statistically significant. Some of these methods, such as family studies, have been going on in the background at molecular genetics labs for many years. Due to the individual limitations of each method (Kerr 2017), labs will rely on multiple lines of evidence when reclassifying variants.
What role does a clinician play in helping to determine the clinical significance of a VUS?
Here are some ways a lab may request your involvement:
- RNA sequencing: obtaining a blood sample, consenting/counseling patients about a new test option
- Family studies: facilitating identification and communication with relatives for family testing
- Reanalysis of data (particularly in exome testing): providing new clinical and/or family history information
Many avenues of investigation are being used by laboratories to help classify genetic variants. One of the areas where you may be playing more of a role is in facilitation of RNA sequencing. RNA sequencing could help a patient like Malik receive a diagnosis. Some day in the near future, you may find yourself ordering RNA sequencing along with DNA analysis when trying to diagnose a genetic condition in your patients.
Resources
Genetic Testing in Pediatric Neurology (free CME/CNE)
Precision Medicine for Your Practice program
Variant classification and reanalysis
Results interpretation tool
References
Abramowicz A, Gos M. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genetics. 2018; 59(3):253-268.
Borras E, Chang K, Pande M, et al. In silico systems biology analysis of variants of uncertain significance in Lynch syndrome supports the prioritization of functional molecular validation. Cancer Prev Res (Phila). 2017; 10(10):580–587.
Frésard L, Smail C, Ferraro NM, et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med. 2019; 25(6):911-919.
Gao M, Zhou H, Skolnick J. Insights into disease-associated mutations in the human proteome through protein structural analysis. Structure. 2015; 23(7):1362-1369.
Ittisoponpisan S, David A. Structural biology helps interpret variants of uncertain significance in genes causing endocrine and metabolic disorders. J Endocr Soc. 2018; 2(8):842-854.
Karam R, Conner B, LaDuca H, et al. Assessment of diagnostic outcomes of RNA genetic testing for hereditary cancer. JAMA Netw Open. 2019; 2(10):e1913900.
Kerr ID, Cox HC, Moyes K, et al. Assessment of in silico protein sequence analysis in the clinical classification of variants in cancer risk genes. J Community Genet. 2017; 8(2):87-95.
Landrith T, Li B, Cass AA, et al. Splicing profile by capture RNA-seq identifies pathogenic germline variants in tumor suppressor genes. Npj Precis Onc. 2020; 4(1).
Lee H, Huang AY, Wang L, et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med. 2020; 22(3):490-499.